Pymatgen and the Materials Project API#

Now that we have learned how to use ASE to build atomic structures, let’s learn about how we can use the Pymatgen and Materials Project API to retrieve known atomic structures from databases and visualize their electronic properties.
Pymatgen is a powerful open-source Python library for materials analysis, designed to interface with electronic structure codes such as VASP and ABINIT. While its functionality is extensive (supporting everything from symmetry analysis to phase diagrams), this tutorial uses Pymatgen primarily as a tool to obtain crystal structures from the Materials Project and convert them to ase.Atoms objects. This interoperability makes it a useful bridge between databases and simulation frameworks.

The Materials Project API allows a user to query information from the Materials Project.
What is an API?
An Application Programming Interface (API) is a set of standardized methods that allows different software programs to talk to one another. In our case, the Materials Project API enables your Python code to access crystal structures, band gaps, and other materials properties directly—without having to manually search and download files from a website.
APIs are especially useful for automating queries. They make getting large amounts of information much more efficient than point-and-click manual access to a web server via an Internet browser.
To access their API, the Materials Project has released a Python client in the mp-api package, which provides a simple interface for querying the Materials Project database and retrieving data over the internet. However, to access the Materials Project, you’ll need an API key, which you can get from your account dashboard after logging into materialsproject.org.
Important
An API key is required to use MPRester.
You must be logged in on materialsproject.org to obtain your API key.
If you do not have a Materials Project account, you can create one for free with a valid email address.
You can obtain your personal API key from your Materials Project Dashboard, or you can get it from the documentation page.
Your API key is a long alphanumeric string (about 30 characters) that you must use every time you wish to query the Materials Project programmatically via the API.
Make sure to never share your API key or accidentally upload code with your API key visible.
Once set up, you can access structured data programmatically—retrieving everything from atomic structures to band gaps and density of states with just a few lines of Python.
Installation#
To use the pymatgen and mp-api packages, we will need to install them using the Python package manager:
pip install pymatgen mp-api
Once installed, you’ll be able to access Pymatgen’s wide suite of materials tools and connect to the Materials Project Database.
Using the MPRester Object#
Let’s get started by connecting the the Materials Project database. First, you will need to copy your API key from your Materials Project Dashboard.
To use your API key in our Python code, we store its value in a string, like this:
# Save your Materials Project API as a string
MP_API_KEY = '---your api key here ----'
After storing your API key in a variable (e.g., MP_API_KEY), you can use it to initialize a connection to the Materials Project with MPRester. This is typically done in a with statement like with MPRester(MP_API_KEY) as mpr:, which opens a session. Inside this block, you can call methods on mpr—such as retrieving structures or material data—using commands like mpr.some_method(). Let’s look at some examples to see how this works in practice.
Get the Crystal Structure of a Specific Material#
In the Materials Project, each material has a unique identifier, known as its Materials Project ID (MPID). When you want information about a single material, it is reasonable to perform a manual search using the Materials Explorer for the material so you can find its MPID. Then, you can use the MPID along with MPRester to automate queries about the material.
As an example, YBa\(_2\)Cu\(_3\)O\(_7\) has mp-20674 as its MPID. We can obtain its crystal structure directly using this MPID as follows:
from mp_api.client import MPRester
import os
import pymatgen as pmg
from ase.io import write
MPID = 'mp-20674' # Materials Project ID number
"""
The 'with ...' statement defines an MPRester code block. Subsequent
indented statements belong to the code block, and the object mpr
may be used within the code block.
"""
with MPRester(MP_API_KEY) as mpr:
# Get the structure for YBa2Cu3O7
structure = mpr.get_structure_by_material_id(MPID)
Show code cell output
The MPRester method get_structure_by_material_id() queries the Materials Project and returns the crystal structure, storing it in an object structure. Having accessed the Materials Project, we no longer require the mpr object. We can now exit the MPRester code block by resetting the indentation.
Next, we provide code to inspect the structure we downloaded. What is its data type? How can we use it?
"""
Reset the indentation (exith the 'with' block), and examine the
structure we obtained.
"""
print('\nWhat is the data type of the structure we obtained?')
print(type(structure))
print('\nWhat is the structure we obtained?')
print(structure)
What is the data type of the structure we obtained?
<class 'pymatgen.core.structure.Structure'>
What is the structure we obtained?
Full Formula (Ba2 Y1 Cu3 O7)
Reduced Formula: Ba2YCu3O7
abc : 3.844668 3.926152 11.823664
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (13)
# SP a b c magmom
--- ---- --- --- -------- --------
0 Ba 0.5 0.5 0.819391 -0
1 Ba 0.5 0.5 0.180609 -0
2 Y 0.5 0.5 0.5 0
3 Cu 0 0 0.646678 -0
4 Cu 0 0 0.353322 -0
5 Cu 0 0 0 0
6 O 0 0.5 0 0
7 O 0.5 0 0.620653 -0
8 O 0.5 0 0.379347 -0
9 O 0 0.5 0.621651 -0
10 O 0 0.5 0.378349 -0
11 O 0 0 0.84082 0
12 O 0 0 0.15918 0
The output of the code above indicates that the data in structure is in a format compatible with pymatgen. pymatgen has a tool to convert the pymatgen structure to an ase object, which we can visualize.
from pymatgen.io.ase import AseAtomsAdaptor as aaa
# convert the structure to an ase.Atoms object
crystal = aaa.get_atoms(structure)
# Make a static visualization
orientation='90x,75y,-9x'
write('YBa2Cu3O7_structure.png', crystal, show_unit_cell=2,
rotation=orientation)
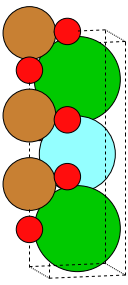
Let’s make an interactive visualization of the crystal:
from ase.visualize import view
# Interactive 3D visualization
view(crystal, viewer='x3d')
Having obtained the crystal structure, we can now use it in a variety of ways:
Use it within an atomistic simulation
Use
ase.io.write()to save the structure in a structure file (*.cif,*.xyz, etc.)
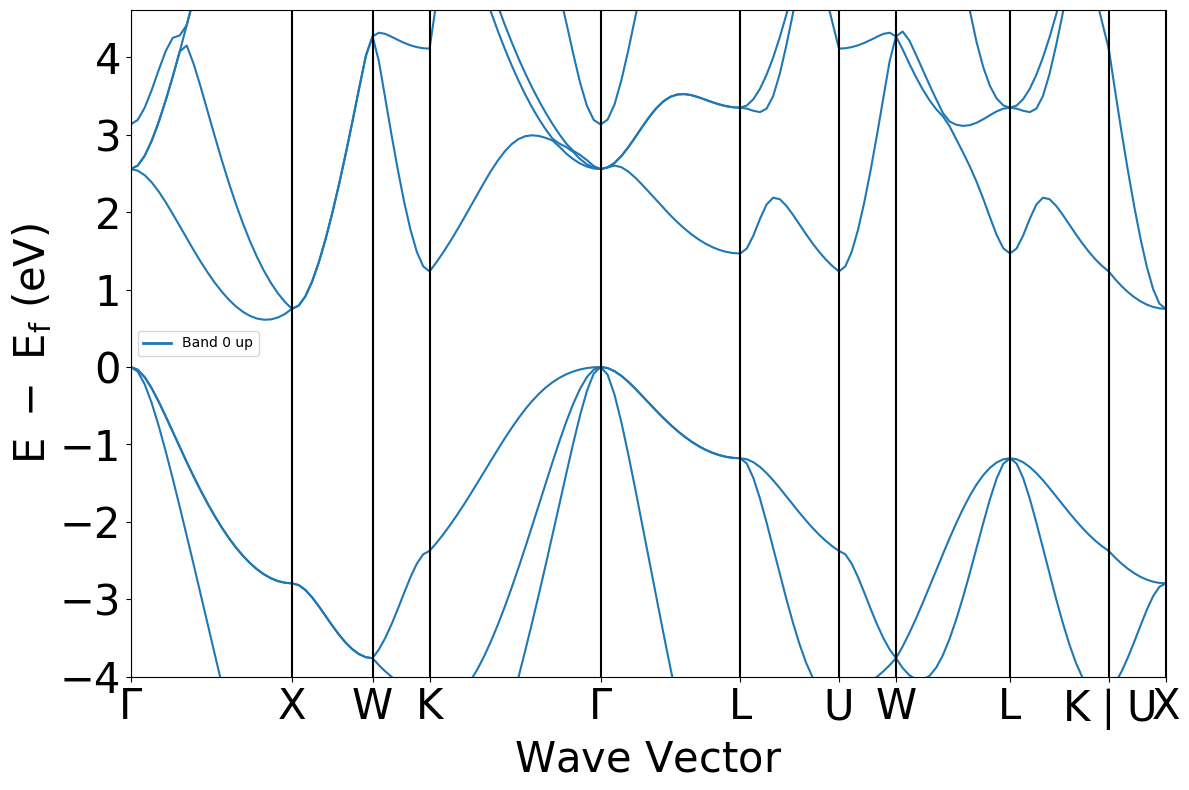
Getting the Band Structure for a Material#
Band structure data provides insight into the electronic properties of materials. By querying with an MPID, you can retrieve a Pymatgen BandStructure object and use BSPlotter to visualize it. This is especially useful for identifying semiconductors, insulators, or materials with interesting electronic behavior like Dirac points or band inversions.
We can use the MPRester class to obtain band structures for a material:
mpid = "mp-149" # this is the MPID for silicon crystal (diamond lattice)
with MPRester(MP_API_KEY) as mpr:
bs = mpr.get_bandstructure_by_material_id("mp-149")
Show code cell output
This returns a Pymatgen band structure object, which can be plotted.
from pymatgen.electronic_structure.plotter import BSPlotter
# plot & show the band structure we obtained
plot = BSPlotter(bs).get_plot()
Searching using MPRester#
Using the MPRester object, Materials Project data can be queried in two ways:
through a specific list of MPID(s), and/or
through property filters (e.g. band gap less than 0.5 eV)
Filters can be applied to find materials that meet specific criteria. The search results return structured documents that contain key properties like chemical formula, symmetry, and electronic structure. You can also customize which properties to return by specifying them in the fields argument.
When querying a list of MPIDs, we use the following syntax:
with MPRester(MP_API_KEY) as mpr:
docs = mpr.materials.summary.search(material_ids=["mp-149", "mp-13", "mp-22526"])
Show code cell output
Here, each material entry in the Materials Project has summary data, and we are simply searching the summary data using mpr.materials.summary.search(). Since we queried for a list of MPIDs, we store in docs a list of “documents” (formally, a list of MPDataDoc objects).
We can now reference an individual document and extract its properties. We’ll use a for loop to list the MPID and chemical formula for each search hit:
print('Our query returned {0} docs.'.format( len(docs) ))
for idx, mat_doc in enumerate(docs):
print('Item {0}: MPID = {1} (formula: {2})'.format(idx,
mat_doc.material_id,
mat_doc.formula_pretty))
Our query returned 3 docs.
Item 0: MPID = mp-149 (formula: Si)
Item 1: MPID = mp-22526 (formula: LiCoO2)
Item 2: MPID = mp-13 (formula: Fe)
What properties ('material_id', 'formula_pretty', etc.) are available for search in the summary data? We can obtain a list of document properties using the following syntax:
print(mpr.materials.summary.available_fields)
['builder_meta', 'nsites', 'elements', 'nelements', 'composition', 'composition_reduced', 'formula_pretty', 'formula_anonymous', 'chemsys', 'volume', 'density', 'density_atomic', 'symmetry', 'property_name', 'material_id', 'deprecated', 'deprecation_reasons', 'last_updated', 'origins', 'warnings', 'structure', 'task_ids', 'uncorrected_energy_per_atom', 'energy_per_atom', 'formation_energy_per_atom', 'energy_above_hull', 'is_stable', 'equilibrium_reaction_energy_per_atom', 'decomposes_to', 'xas', 'grain_boundaries', 'band_gap', 'cbm', 'vbm', 'efermi', 'is_gap_direct', 'is_metal', 'es_source_calc_id', 'bandstructure', 'dos', 'dos_energy_up', 'dos_energy_down', 'is_magnetic', 'ordering', 'total_magnetization', 'total_magnetization_normalized_vol', 'total_magnetization_normalized_formula_units', 'num_magnetic_sites', 'num_unique_magnetic_sites', 'types_of_magnetic_species', 'bulk_modulus', 'shear_modulus', 'universal_anisotropy', 'homogeneous_poisson', 'e_total', 'e_ionic', 'e_electronic', 'n', 'e_ij_max', 'weighted_surface_energy_EV_PER_ANG2', 'weighted_surface_energy', 'weighted_work_function', 'surface_anisotropy', 'shape_factor', 'has_reconstructed', 'possible_species', 'has_props', 'theoretical', 'database_IDs']
Next, we query using property filters. We apply the following filters:
Materials containing Si and O
Materials with a band gap no greater than 1.0 eV but no less than 0.5 eV
Instead of all available summary fields, we’ll only ask for a few:
"material_id","formula_pretty","band_gap".
with MPRester(MP_API_KEY) as mpr:
docs = mpr.materials.summary.search(
elements=["Si", "O"],
band_gap=(0.5, 0.75),
fields=[
"material_id",
"formula_pretty",
"band_gap"
])
example_doc = docs[0]
# initial_structures = example_doc.initial_structures
Show code cell output
To see what our search turned up, we can use some simple code, like this. We first find out how many hits our query returned using len(docs), and then we print only the first N hits, where we set N = 10.
N = 10
print('Our query returned {0} docs.'.format( len(docs) ))
print(f'Printing only the first {N} results:')
for idx in range(0,N):
mat_doc = docs[idx]
print('Item {0}: MPID = {1} ({2}), band gap = {3:6.4f} eV'.format(idx,
mat_doc.material_id,
mat_doc.formula_pretty,
mat_doc.band_gap))
Our query returned 244 docs.
Printing only the first 10 results:
Item 0: MPID = mp-640917 (SiO2), band gap = 0.6554 eV
Item 1: MPID = mp-554498 (SiO2), band gap = 0.5051 eV
Item 2: MPID = mp-1192103 (FeSiO4), band gap = 0.6887 eV
Item 3: MPID = mp-1199561 (FeSiO3), band gap = 0.7031 eV
Item 4: MPID = mp-725556 (Pu2SiO10), band gap = 0.7104 eV
Item 5: MPID = mp-28195 (SiAg2O3), band gap = 0.6373 eV
Item 6: MPID = mp-1016879 (CdSiO3), band gap = 0.6506 eV
Item 7: MPID = mp-754234 (V2SiO4), band gap = 0.6219 eV
Item 8: MPID = mp-1143241 (CrSiO4), band gap = 0.7340 eV
Item 9: MPID = mp-1205049 (SiAg5O4), band gap = 0.7138 eV
Exercises#
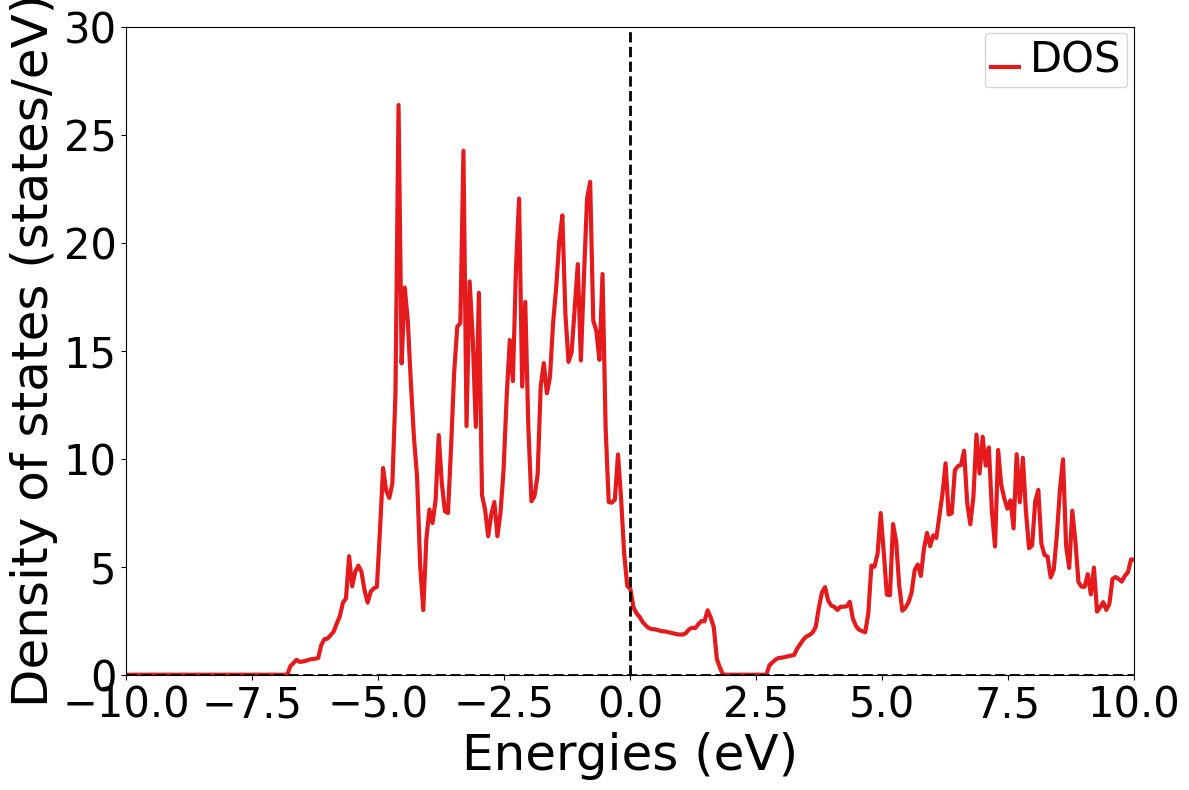
Exercise 1: Visualize the DOS of YBa\(_2\)Cu\(_3\)O\(_7\)
Let’s get some hands-on experience with accessing and plotting electronic structure data, a critical step in evaluating materials for electronic applications in real-world technologies.
First, obtain the electronic density of states (DOS) for YBa\(_2\)Cu\(_3\)O\(_7\). Then plot it using the functionality available in pymatgen.
Hints:
To find the code to obtain the DOS, a Google search such as “MPRester DOS example” may help, or perhaps you can try asking an AI chatbot.
Once you have obtained a DOS and saved it as, say,
some_DOS, you can plot it using code such as this:
from pymatgen.electronic_structure.plotter import DosPlotter
import matplotlib.pyplot as plt
with MPRester(MP_API_KEY) as mpr:
some_DOS = <your code to get the DOS>
# obtain a DosPlotter object
Plotter = DosPlotter()
# add the DOS to the plotter
Plotter.add_dos('DOS', some_DOS)
"""
Choose appropriate numbers for:
E_lo and E_hi, the upper and lower limits
of the domain for your DOS plot.
MaxDensity, the upper limit for the range of
your DOS plot.
This may require some trial and error!
"""
Plotter.get_plot(xlim=(E_lo, E_hi), ylim=(0, MaxDensity))
plt.show()
Solutions#
Exercise 1: YBCO DOS#
Show code cell content
from mp_api.client import MPRester # client for Materials Project
from pymatgen.electronic_structure.plotter import DosPlotter
import matplotlib.pyplot as plt
YBCO = 'mp-20674' # Materials Project ID number
with MPRester(MP_API_KEY) as mpr:
YBCO_DOS = mpr.get_dos_by_material_id(YBCO)
print(YBCO_DOS)
# plot & show DOS we obtained
Plotter = DosPlotter()
Plotter.add_dos('DOS', YBCO_DOS)
Plotter.get_plot(xlim=(-10, 10), ylim=(0, 30))
plt.show()
Complete DOS for Full Formula (Ba2 Y1 Cu3 O7)
Reduced Formula: Ba2YCu3O7
abc : 3.844668 3.926152 11.823664
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (13)
# SP a b c
--- ---- --- --- --------
0 Ba 0.5 0.5 0.819391
1 Ba 0.5 0.5 0.180609
2 Y 0.5 0.5 0.5
3 Cu 0 0 0.646678
4 Cu 0 0 0.353322
5 Cu 0 0 0
6 O 0 0.5 0
7 O 0.5 0 0.620653
8 O 0.5 0 0.379347
9 O 0 0.5 0.621651
10 O 0 0.5 0.378349
11 O 0 0 0.84082
12 O 0 0 0.15918